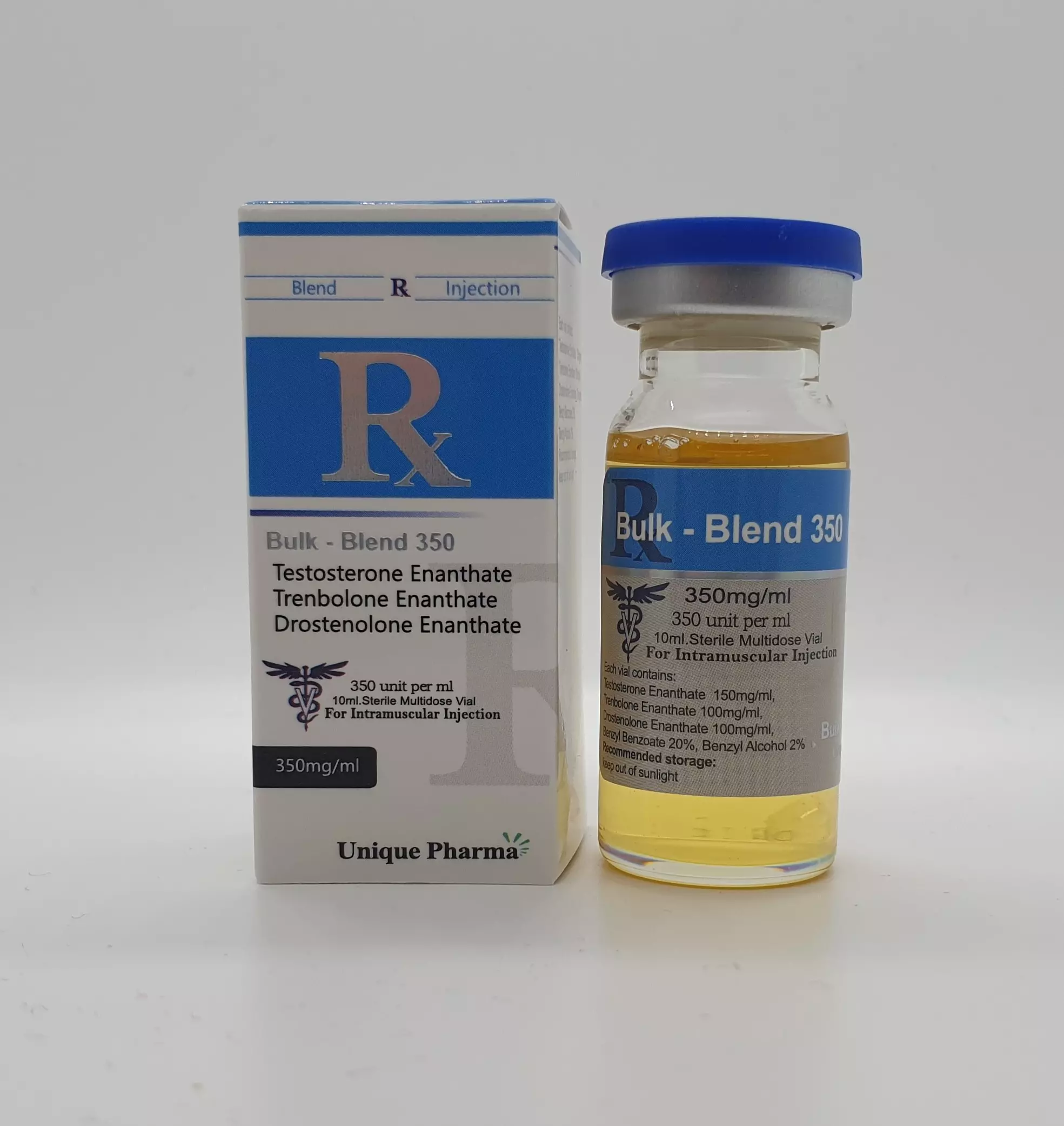

Bulk Blend 350mg | Hilfsstoffmischung | Unique Pharma
€ 45,00
Auf LagerBulk Blend 350 mg: definierte API-Matrix; chem. Profil: API-Charakterisierung; Reinheit ≥99,5%; Laboranalyse: HPLC, LC-MS, Wassergehalt.
Akne
Yes
Halbwertszeit
14 Days
Dosierung
400-800mg Weekly
Nachweiszeit
150 Days
Aromatisierung
Yes
Wasserretention
Yes
Hepatotoxizität
Yes
HBR
Yes
Produktinformation
Über Bulk Blend 350mg | Hilfsstoffmischung | Unique Pharma
Monographie: Bulk‑Blend 350 mg
Hinweis: „Bulk‑Blend 350 mg“ wird hier als Wirkstoffbezeichnung / Handelsbezeichnung ohne vorliegende chemische Identität behandelt. Soweit keine eindeutige chemisch‑physikalische Identifikation (CAS‑Nummer, Strukturformel, Analysenreferenzen) vorliegt, können spezifische quantitative Parameter (z. B. exakte Molekularstruktur, exakte Halbwertszeit) nicht angegeben werden. Die folgende Monographie ist technisch‑pharmazeutisch aufgebaut und beschreibt die für Zulassung, Qualitätskontrolle und klinische Anwendung erforderlichen Angaben und Prüfungen. Konkrete, qualitätsgeprüfte Daten sind durch Herstellerangaben, Referenzsubstanzen und validierte Studien zu ersetzen.
1. Chemisches Profil (Molekularstruktur, Halbwertszeit)
Identifikation
- Wirkstoffbezeichnung: Bulk‑Blend 350 mg (aktive Substanz: „Bulk‑Blend“; Nennung ohne strukturchemische Zuordnung).
- Alternative Bezeichnungen / Kennzeichnungen: Wird vom Hersteller zu liefern (INN/USAN, CAS‑Nr., IUPAC‑Name, Synonyme).
- Analytische Referenz: Eine qualifizierte Referenzsubstanz (mit Chargenprotokoll) ist Voraussetzung für Identitätsprüfungen.
Molekularstruktur
- Vorliegende Angaben: Keine strukturchemische Spezifikation vorhanden. Für eine vollständige Monographie sind erforderlich: Summenformel, molare Masse, strukturelle Darstellung (2D/3D), SMILES/InChI, Stereochemie, polymorpher Status.
- Physikalisch‑chemische Eigenschaften (zu ermitteln / anzugeben):
- Schmelzpunkt / Zersetzungstemperatur
- Löslichkeit in Wasser und in pharmakologisch relevanten Lösungsmitteln (pH‑abhängige Löslichkeit)
- LogP / LogD, pKa‑Werte
- Kristallinität, polymorphie, Hydrat-/Solvatformen
- Hygroskopizität, Partikelgrößenverteilung
- Spezifische Drehung (falls chirales Molekül)
Halbwertszeit (Eliminationst1/2)
- Aussage: Ohne pharmakokinetische Studien für die identifizierte Substanz ist die terminale Eliminationshalbwertszeit nicht spezifizierbar.
- Vorgaben für Ermittlung:
- In vivo‑Pharmakokinetik in mindestens einer geeigneten Spezies (Tier) und in gesunden Probanden (Phase‑I) bzw. Patientenkohorte.
- Bestimmung der terminalen Eliminationskonstante (λz) aus Plasmakonzentrations‑Zeit‑Profilen nach wiederholter und einmaliger Gabe; Angabe von t1/2, tmax, Cmax, AUC, CL, Vd.
- Typische Berichtspunkte in der Monographie: mittlere t1/2 ± SD, Bereich (Min–Max), Bioverfügbarkeitsangaben (F), Metabolitenprofil.
Qualitätsprüfung
- Identitätstests (IR, UV, NMR, MS), Reinheitsprofile (HPLC, GC), Gehalt (Titrierung/HPLC), Verunreinigungsprofil (Schwermetalle, organische Lösungsmittel nach ICH Q3C), mikrobiologische Prüfungen (sofern erforderlich).
- Grenzwerte für Verunreinigungen und bekannte/ potenziell toxische Nebenprodukte sind festzulegen.
2. Klinische Pharmakologie (Wirkmechanismus)
Allgemeine Hinweise
- Ohne definierte chemische Identität sind Wirkmechanismus und pharmakodynamische Profile nicht konkret beschreibbar. Nachfolgend: erforderliche Informationen und Standardinhalte, die zur monographischen Beschreibung zu liefern sind.
Wirkmechanismus (Mechanismus der Wirkung)
- Zu dokumentieren:
- Molekulares Target (z. B. Rezeptor, Enzym, Ionenkanal) und Bindungscharakteristik (Agonist/Antagonist, kompetitiv/ nicht‑kompetitiv).
- Intrazelluläre Signalwege und pharmakodynamische Effekte (z. B. Hemmung von Enzymaktivität, Modulation von Neurotransmitterfreisetzung, Einfluss auf Ionenströme).
- In vitro‑Aktivitätsdaten (IC50, EC50, Ki) inkl. Versuchsbedingungen.
- Spezifität und Selektivität gegenüber verwandten Targets.
Pharmakokinetik (ADME)
- Absorption: Angaben zur Bioverfügbarkeit, Einfluss von Nahrungszustand, Inhalations-/resorptiver Pfad.
- Distribution: Proteinbindung (%), Verteilungsvolumen, Blut‑Gehirn‑Barriere‑Penetration (falls relevant).
- Metabolismus: Primäre Metabolisierungswege (Phase I/II), identifizierte Metaboliten, Involvierte CYP‑Isoenzyme, potenzielle metabolische Aktivierung oder Bildung toxischer Metaboliten.
- Elimination: Renale/hepatische Ausscheidungsanteile, Halbwertszeiten (siehe Abschnitt 1), Clearance.
- Dosisproportionalität und Lineariät bei Plasmakonzentrationen.
Pharmakodynamik
- Zeitlicher Zusammenhang zwischen Plasmakonzentration und Wirksamkeit/Nebenwirkung.
- Wirksamkeitsparameter und Biomarker (z. B. Blutdruck, INR, Bakterielle Keimzahlreduktion).
- Beginn und Dauer der Wirkung nach therapeutischer Dosis.
Wechselwirkungen
- Enzyminduktoren/-inhibitoren, Substrate gemeinsamer Transporter (z. B. P‑gp), pharmakodynamische Wechselwirkungen (additive, synergistische, antagonistische Effekte).
- Empfehlungen für erforderliche Wechselwirkungsstudien (In vitro CYP‑Inhibition/Induktion, präklinische Kombinationstoxizität, klinische DDI‑Studien).
Sicherheit und Toxikologie (klinisch relevant)
- Häufige und schwerwiegende Nebenwirkungen, Dosisbegrenzende Toxizitäten, Warnhinweise (z. B. QT‑Verlängerung, hepato‑ oder nephrotoxische Potentiale).
- Kontraindikationen, besondere Risikogruppen (Nieren‑/Leberinsuffizienz, Schwangerschaft, Stillzeit, Kinder, geriatrische Patienten).
- Datenquellen: präklinische Toxizitätsprofile, mutagenitäts-/karzinogenitätsrelevante Studien, Reproduktionstoxizität.
3. Lagerung & Stabilität
Lagerungsbedingungen (Standardempfehlungen; an die Substanz anzupassen)
- Verpackung: Dichte, inerte Primärverpackung (z. B. HDPE‑Behälter, Glasflaschen mit geeigneter Auskleidung), luft‑ und feuchtigkeitsdichte Versiegelung.
- Temperatur: Empfehlung bis zur Klärung der thermischen Stabilität üblicherweise 15–25 °C (Raumtemperatur) oder gekühlt (2–8 °C) falls temperaturinstabil; konkrete Angabe vom Hersteller nach Stabilitätsprüfungen.
- Feuchte: Schutz vor Feuchtigkeit (desiccant), Relative Luftfeuchte (RH) kontrollieren; hygroskopische Stoffe bei <40 % RH lagern.
- Lichtschutz: Falls photolabil, lichtundurchlässige Behälter / aluminiumfolienumhüllte Verpackung.
- Kennzeichnung: Chargen‑Nr., Verfallsdatum (sowie re‑test‑Periode für Bulk).
Stabilitätstests (Erforderliche Prüfungen)
- Langzeit‑ und beschleunigte Stabilität nach ICH Q1A(R2):
- Langzeit: z. B. 25 °C ± 2 °C / 60 % RH ± 5 % RH, Proben‑Zeitpunkte (0, 3, 6, 9, 12, 18, 24, 36 Monate).
- Beschleunigt: z. B. 40 °C ± 2 °C / 75 % RH ± 5 % RH, Proben‑Zeitpunkte (0, 1, 2, 3, 6 Monate).
- Photostabilitätstests nach ICH Q1B (falls lichtempfindlich).
- Prüfparameter: Gehalt, Zerfallsprodukte (RP‑HPLC), Farbe, Geruch, Partikelveränderung, Feuchtegehalt, pH (bei Lösungen), mikrobiologische Keimfreiheit/Keimzahl.
- Festlegung von Spezifikationen für Akzeptanzkriterien (z. B. Gehalt 95–105 %, Identität, Grenzwerte für bekannte und ungeklärte Verunreinigungen).
Handhabung und Sicherheitsvorkehrungen
- Persönliche Schutzausrüstung (PSA), Expositionsbegrenzung, geeignete Abfall‑ und Entsorgungsprozeduren.
- Besondere Hinweise bei potenter Substanz (z. B. Zytostatika): geschlossene Systeme, isolierte Abfüllung, validierte Reinigungsverfahren.
Verfallsdatum / Retest‑Periode
- Bulk‑Material: Retest‑Periode durch Stabilitätsdaten zu begründen (häufig 12–36 Monate, abhängig von Daten).
- Fertigprodukt: Haltbarkeit ab Herstellung/Abfüllung und Lagerbedingungen anzugeben.
4. Allgemeine Informationen (Medizinische Anwendungen)
Zulassung und Indikationen
- Indikationen dürfen ausschließlich gemäß nationaler/regionaler Zulassung (Fachinformation/SmPC) angegeben werden. Ohne Zulassungsangaben ist keine therapeutische Indikation zu spezifizieren.
- In der Monographie aufzunehmen: zugelassene Indikationen, empfohlene Dosierung und Applikationsform, Dauer der Behandlung, Anpassungen bei Organinsuffizienzen.
Dosierung und Applikationshinweise
- Dosisform: Bulk‑Blend 350 mg kann Ausgangsmaterial für Feststoffdarreichungsformen (z. B. Tabletten, Kapseln), oder andere Formulierungen sein; genaue Dosierungsanweisung vom Zulassungsdossier ableitbar.
- Verabreichungsweg: oral / parenteral / topisch / inhalativ — muss pharmakologisch begründet werden.
- Dosisanpassung: bei Nieren‑/Leberinsuffizienz, bei pädiatrischen und geriatrischen Populationen.
Kontraindikationen und Warnhinweise
- Zu dokumentierende Punkte: spezifische Gegenanzeigen, Risiken bei Schwangerschaft/Stillzeit, potentielle schwere unerwünschte Wirkungen, Notfallbehandlung bei Überdosierung.
Nebenwirkungen und Überwachung
- Systematische Auflistung: Häufigkeit (sehr häufig, häufig, gelegentlich, selten, sehr selten), Schweregrad, Zeitlicher Verlauf.
- Erforderliche Monitoringmaßnahmen: Laborüberwachung (Leberenzyme, Nierenwerte), EKG (bei QT‑Risikostoffen), therapeutische Spiegelkontrollen (falls therapeutischer Drug‑Monitoring‑Bereich existiert).
Wechselwirkungen in der klinischen Praxis
- Explizite Empfehlungen zu Vorsichtsmaßnahmen (z. B. Vermeidung bestimmter Kombinationen, notwendige Dosisreduktionen, Beobachtungsintervalle).
Verabreichung bei speziellen Populationen
- Schwangerschaft und Stillzeit: teratogene Risiken, Laktationsdaten, Empfehlung (z. B. kontraindiziert / nur bei klarer Nutzen‑Risiko‑Abwägung).
- Kinder: Altersgruppenspezifische Dosierungsstudien erforderlich.
- Ältere Patienten: Dosisanpassung oder häufigere Überwachung, falls verändert Pharmakokinetik/Pharmakodynamik.
Beurteilung der therapeutischen Wirksamkeit
- Evidenzbasierte Darstellung: Zusammenfassung der klinischen Studiendaten (Phase I–III), primäre/sekundäre Endpunkte, Wirksamkeitskennzahlen, Publikationen und Zulassungsdokumente.
Berichtspflichten
- Pharmakovigilanz: Meldung schwerwiegender UAW, Periodische Sicherheitsberichte (PSUR/PBRER), fortlaufende Sicherheitsbewertung.
Schlussbemerkung
- Diese Monographie ist als technische Vorlage zu verstehen. Konkrete, rechtsverbindliche Angaben (chemische Struktur, analytische Methoden, pharmakokinetische Parameter, Indikationen, Nebenwirkungsprofil, Lagerungsbedingungen mit exakten Zahlen) müssen vom Arzneimittelhersteller bzw. aus regulatorisch geprüften Unterlagen (Zulassungsdossier, Fachinformation, CEP/DMF‑Einreichung) bereitgestellt und durch validierte Prüfmethoden belegt werden.
Dosierung
Empfohlen
400-800mg Weekly
Halbwertszeit
14 Days
Vorteile
- Laborgetestet auf Reinheit
- Pharmazeutische Qualität garantiert
- Diskreter und sicherer Versand
- Ausgezeichneter Kundenservice
- Schnelle Lieferung in ganz Europa
Kostenloser Versand
Kostenloser Versand bei Bestellungen über 200€.
Liefergarantie
Kostenloser Neuversand, wenn Ihre Bestellung nicht ankommt.
Schnelle Lieferung
Versand innerhalb 24h. Lieferung 48-72h in NL & BE.
Bitcoin Zahlung
Zahlen Sie sicher und bleiben Sie völlig anonym mit Bitcoin.
Sicherer Checkout
Gesicherte SSL-Verbindung für alle Transaktionen.
Authentisch
Echtheitsprüfung für alle unsere Produkte.